Eine pilot-gen-Therapie für Sichel-Zelle Krankheit, Wiederherstellung der Patienten’ Fähigkeit, das fetale Hämoglobin, produziert gute Ergebnisse in den ersten drei Patienten zu erhalten. Die Ermittler in Boston Children ‘ s Hospital berichtet, die Ergebnisse Ihrer Laufenden klinischen Studie in dieser Woche an der amerikanischen Gesellschaft für Hämatologie (ASH) annual meeting.
Die drei Erwachsenen Patienten behandelt, im Boston Children ‘ s, wurden nun folgte für 8, 10 und 18 Monaten, beziehungsweise. Keiner hat gezeigt, unerwünschte Ereignisse aus der Behandlung. Und alle Indikatoren deuten darauf hin, dass die Gentherapie hat sich Ihrer “angenommen” und das sickling von roten Blutkörperchen reduziert wurde. Der Prozess hat jetzt begonnen, sich zu registrieren Jugendlichen mit Sichelzellanämie.
Wiederherstellung des fetalen Hämoglobins
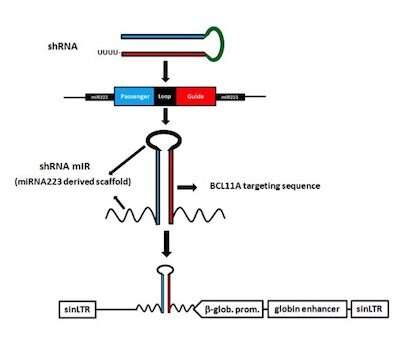
Die gen-Therapie, Zeichnung nach rund 70 Jahren der Forschung, ist entworfen, um die Menschen zu machen, die fötale form des Hämoglobins, die normalerweise nicht nach der Geburt. Die Mutationen verursacht, die sichelzellkrankheit, haben keinen Einfluss auf das fetale Hämoglobin, also die roten Blutkörperchen, die zu tragen es zu erwarten wäre, nicht zu Sichel. Die Behandlung schweigen BCL11A, das gen, das heruntergefahren fetalen Hämoglobin-Produktion. In so doing, beide neu gestartet, das fetale Hämoglobin-Produktion und unterdrückt die Produktion des Erwachsenen, sickling form von Hämoglobin.
Tests auf die drei Patienten zeigten, dass die Blut-Stammzellen modifiziert mit der Gentherapie-Vektor (ein genetisch verändertes virus mit erweiterten Sicherheits-features) erfolgreich engrafted bei Patienten, Knochenmark-und bleiben in Ihrer Durchblutung. Wie erhofft, werden die Ebenen des BCL11A-protein signifikant reduziert bei Patienten’ roten Blutkörperchen-Vorstufen, blieb aber normal in der anderen Blutzellen.
Genauso, wie die Forscher gehofft hatten, alle drei Patienten zeigten einen signifikanten Anstieg der fetalen Hämoglobins. Eine Fortsetzung zu get red-blood-cell-Transfusionen aufgrund von bereits bestehenden Blutgefäßen zu Schäden im Gehirn, eine bekannte Komplikation der Sichelzellanämie. Aber bei den anderen beiden Patienten, die roten Blut-Zellen mit fetalem Hämoglobin aus etwa 70 Prozent aller roten Blutkörperchen. In beiden fetalen Hämoglobin-Spiegel wurden gedacht, um hoch genug sein, um zu verhindern, dass sickling. Zusätzlich werden die Patienten nicht mehr zeigen, die Anämie bei Patienten mit Sichelzellanämie.
“Das gen-Therapie-Behandlung wurde sicher und gut verträglich”, sagt Erica Esrick, MD, co-principal investigator der klinischen Studie und ein pädiatrischer Hämatologe an der Boston Kinder. “Wir sind erfreut zu sehen, einen erheblichen und stabilen Anstieg der gesamten fetalen Hämoglobins und der Menge des fetalen Hämoglobins pro Zelle.”
Entwerfen Sie eine Sichel Zell-Gentherapie-Vektor
Es ist bereits bekannt seit den 1970er-Jahren, dass einige Menschen mit sichelzellen-mutation halten produzierenden fetalen Hämoglobins und milder Erkrankung. Vijay Sankaran, MD, Ph. D. und Stuart Orkin, MD von Boston Children ‘ s konzentrierte sich auf BCL11A in 2008. In einer Studie aus dem Jahr 2011, Orkin s team unterdrückt BCL11A in einem Maus-Modell und erfolgreich in umgekehrter Sichel-Zelle Krankheit.

Für die klinische Studie, die ein team unter der Leitung von David A. Williams, MD, Boston Children ‘ s Chief Scientific Officer, ging zum Ingenieur eine optimierte Gentherapie Vektor zu packen. Es unterdrückt BCL11A, aber nur in Vorstufen der roten Blutkörperchen.
“Unser Ansatz ist, kehrt der physiologischen Hämoglobin-Schalter, erhöhen gleichzeitig das fetale Hämoglobin und verringern direkt sickling Hämoglobin”, sagt Williams, der auch der sponsor der klinischen Studie. “Weitere Studien sind das hinzufügen von Genen, die Kodieren fetalen Hämoglobins oder korrigiert, nicht-sickling Erwachsenen Hämoglobin, ohne direkt abnehmende expression der Sichel Hämoglobin-gene. Sagen wir, diese Strategie ist ein effizienter und effektiver Weg, um zu verringern oder sogar beseitigen die sickling Zellen.”
Manny ‘ s Geschichte
Manny Johnson aus Boston, 22, war der erste patient behandelt; seine frühen Ergebnisse wurden auf der ASH im letzten Jahr. Seine Sichel Zelle Krankheit war überaus schwer, und hatte ihm zu haben monatliche Bluttransfusionen. Jetzt, mehr als 18 Monate später, er blieb symptomfrei und nicht benötigte Transfusionen.






